Epidemiology
History
- Historical Term for CF: Mucoviscidosis
- First Historical Connection Between Lung Disease and Sweat Abnormality in CF: there was the first identification of a connection between presence of lung disease in subset of patients and a sweat abnormality during the 1940’s heat wave in Manhattan
- Death of a number of these patients was likely due to high degree of salt loss during the heat wave
Frequency
- Prevalence: CF is the most common lethal genetic syndrome in whites
- Occurs in 1/2500 Caucasians in US
- Occurs in 1/17,000 blacks in US
- Most cases detected by newborn screening in California are in Hispanics
- Rare in American Indians, black Africans, and Asians
Genetics
- Inheritance: autosomal recessive
- Gene Carrier State
- 2-5% of Caucasians are clinically asymptomatic carriers of gene
- There is some reproductive advantage for the carrier state: possible historically reduced diarrheal response to cholera
Physiology
Genetics of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)
- Gene for CFTR is Located on Chromosome 7: gene codes for the 1480 amino acid CFTR protein
Normal Function of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Protein
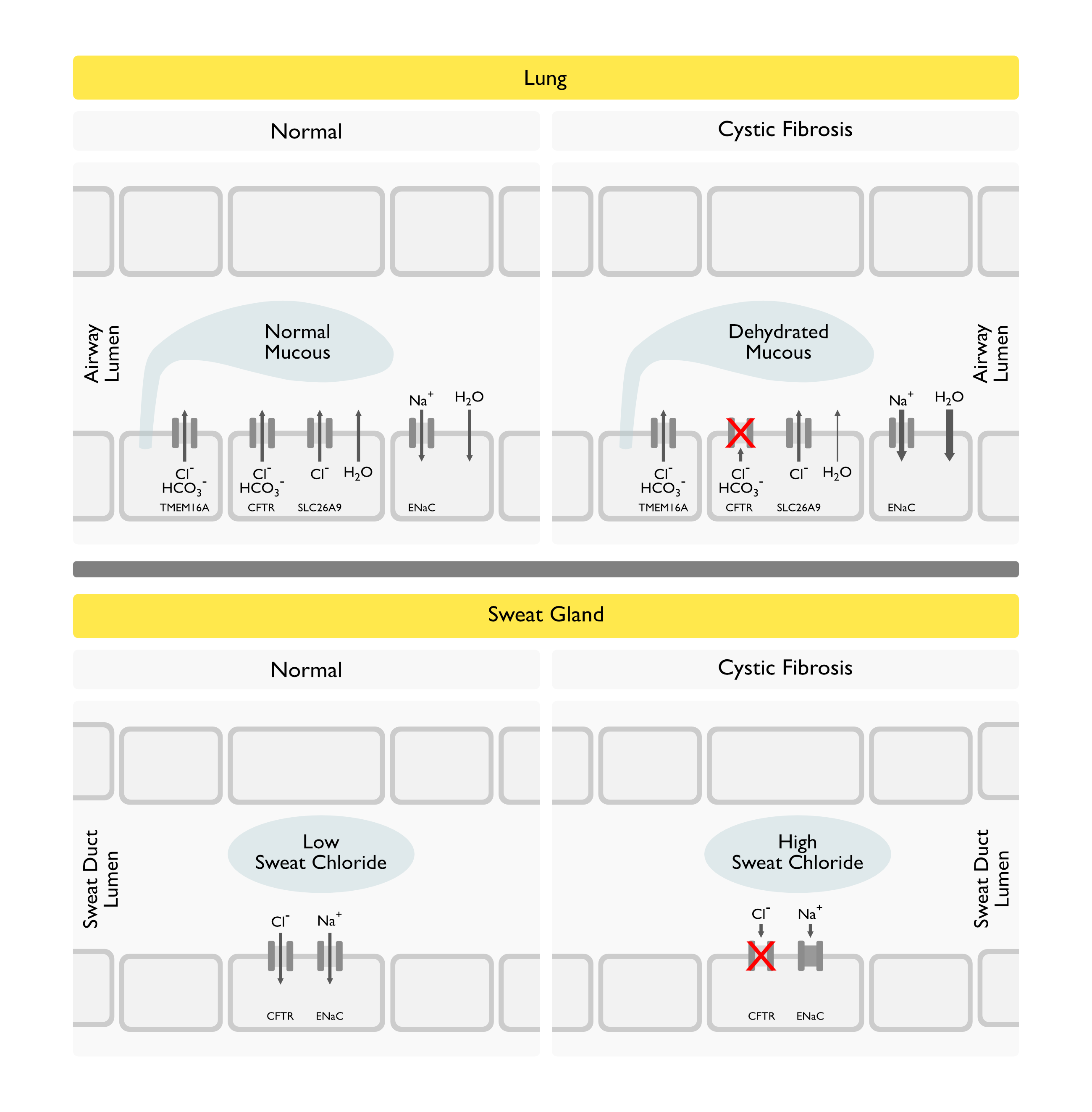
- CFTR Functions as CAMP-Regulated Anion Channel on the Apical Membrane of Fluid/Electrolyte-Transporting Epithelial Cells in Multiple Organ Systems
- CFTR: located in the apical membrane (of predominantly epithelial cells)/ acts as chloride channel, modulates other membrane channels as well as exocytosis and endocytosis, trans-=ports ATP/ regulated by CAMP-dep. kinase and C-kinase
- Airway epithelial CFTR in CF does not respond to ß-agonists (increases CAMP)
- Airway (but not glandular) epithelial CFTR in CF responds to increased intracellular Ca+
- Na+ absorption by airway epithelia in CF is worsened by ß-agonists (and other agents that increase CAMP)
Mutations of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)
- Known Mutations Account for 90% of CF Cases: there are currently >1900 mutations known
- F508del Mutation: mutation deletes phenylalanine from the CFTR protein
- Most common mutation (present in 68% of CF cases)
- Variability in Clinical Presentation: can be partly attributed to genotype
- Example: exocrine pancreatic insufficiency and possibly more severe lung disease are present in homozygous F508del cases
Types of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Mutations
- Class I Mutations: mutations resulting in partial or total absence of CFTR protein expression
- Includes the nonsense mutations and that which produce a premature stop codon
- Characterized by loss of conductance of chloride channel in affected epithelia
- Class II Mutations: mutations resulting in altered cellular maturation of CFTR protein
- Protein is absent from plasma membrane or present in a very small quantity
- Class II mutations represent the majority of CF alleles (DF508)
- Class III Mutations: mutations disturbing the regulation of the chloride channel
- These mutations are frequently siutated in the ATP binding domain (NBF1, NBF2)
- Class IV Mutations: mutations altering conduction of the chloride channel
- Missense mutations affecting segments of the membrane-spanning domain (which form an ionic pore) produce a correctly positioned CFTR protein that have cAMP-depedent chloride channel activity, but which has decreased ion flux and modified selelctivity (R117H)
- Class V Mutations: mutations altering stability of CFTR mRNA
- Class VI Mutations: mutations altering the stability of the mature CFTR protein
Physiologic Consequences of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Protein Dysfunction
Impairment of Epithelial Chloride Transport: resulting in increased NaCl loss through sweat
- CF patients have higher salt (NaCl) losses, but do not “overheat” more readily than normal patients
-
Lung mucosal dysfunction: affects airway but not alveolar epithelia/ secretions (with mucin glycoproteins, secreted in response to proteases and mediators) resist clearance and lead to bacterial infection (and neutrophil chemotaxis/ release of proteolytic enzymes with impaired ciliary function/ further hypersecretion)
Role of lung infection (predominantly S. Aureus and P. Aeruginosa, possibly due to adherence): due to impaired mucociliary clearance (although decreased clearance from central airways is minimal)/ probable glandular cellular dysfunction/ decreased lymphocyte proliferation in response to Pseudomonas (in later stages)/ impaired opsonin-mediated phagocytic activity (possibly due to excessive IgG2 in milieu/ excessive proteases altering complement)/ possibly linoleic acid deficiency/ Pseudomonas antigens induce IL-8 production by human airway epithelial cells and monocytes
- CF cases with decreased IgG have less severe lung disease (Matthews, 1980), suggests sever-ity may be related to hyperimmune response
Pathology
-
General pathology: dilation and hypertrophy of mucous glands/ goblet cell hyperplasia/ excessive mucous secretions in GI and respiratory tracts/ impaired mucous transport in trachea, sinuses, salivary glands, biliary system, apocrine sweat glands (but not eccrine sweat glands)/ pancreatic acinar loss with relative sparing of Islets (for extended periods), pancreatic destruction (90% of cases at autopsy)/ focal biliary cirrhosis (25% of cases): normal parenchyma separated by fibrous bands/ hypoplastic gallbladder containing gelatinous material/ minimal small intestinal mucosal changes/ rectal mucosal goblet cell hyperplasia (inconsistent though)/ destroyed vas deferens, epididymis, seminal vesicles/ plugging of cervix with endocervicitis/ normal parotid and sweat glands, abnormal mucous-secreting salivary glands
-
Lung/respiratory pathology (lungs are normal at birth but undergo changes over time): excessive neutrophils, neutrophil proteases (which cause hypersecretion) and DNA in sputum/ sputum possesses IL-8 mediated neutrophil chemotactic ability/ plugging of proximal airways, bronchioles/ small amounts of destructive emphy-sematous changes and sometimes bullae (especially in patients that live to age 20-30)/ absence of extensive alveo-lar wall destruction/ sometimes DAD, interstitial pneum-onitis with organ-izing pneumonia, interstitial cysts, or UIP patterns are seen/ peribronchi-olar and peribron-chial fibrosis (may cause restriction in end-stage cases)/ enlarged bronchial arteries/ chronic inflammation of paranasal sinuses and nasal passages, nasal polyps
Diagnosis
Arterial Blood Gas (ABG) (see Arterial Blood Gas, [[Arterial Blood Gas]])
- Daytime Hypoxemia
- Nocturnal Hypoxemia: worst during REM, possibly due to decreased FRC
- Hypercapnia: seen only in late stages, when FEV1 falls below 30% predicted
Sputum Gram Stain and Culture
General Information
- Oropharyngeal Swabs Correlate with Bronchoalveolar Lavage When Positive
Common Organisms Isolated from Respiratory Tract in Cystic Fibrosis (All Ages, From 2013 Cystic Fibrosis Registry)
- Aspergillus (see Aspergillus, [[Aspergillus]]): isolated from 11.9% of patients
- Burkholderia Cepacia Complex (Burkholderia Cenocepacia, Burkholderia Multivorans, etc) (see Burkholderia Cepacia, [[Burkholderia Cepacia]]): isolated in 2% of patients
- Haemophilus Influenzae (see Haemophilus Influenzae, [[Haemophilus Influenzae]]): isolated in 11% of patients
- Seen early in course
- Non-Tuberculous Mycobacteria: isolated in 71% of patients who are at least 12 y/o and are taking a macrolide
- Mycobacterium Abscessus (see Mycobacterium Abscessus, [[Mycobacterium Abscessus]])
- Mycobacterium Avium Complex (MAC) (see Mycobacterium Avium Complex, [[Mycobacterium Avium Complex]])
- Mycobacterium Chelonae (see Mycobacterium Chelonae, [[Mycobacterium Chelonae]])
- Other Mycobacteria
- Pseudomonas Aeruginosa (see Pseudomonas Aeruginosa, [[Pseudomonas Aeruginosa]]): isolated from 46% of patients
- Multiple Drug-Resistant Pseudomonas: isolated from 16% of patients
- Prevalence Increases with Age: overall prevalence reaches 75% in adult CF patients
- May be mucoid
- Recovery in a young patient is suggestive of CF
- Staphylococcus Aureus (see Staphylococcus Aureus, [[Staphylococcus Aureus]]): isolated from 70% of patients
- Methicillin-Resistant Staphylococcus Aureus (MRSA): isolated from 52% of patients
- Stenotrophomonas Maltophilia (see Stenotrophomonas Maltophilia, [[Stenotrophomonas Maltophilia]]): isolated from 14% of patients
Other Organisms
- Alcaligenes (Achromobacter) Xylosoxidans (see Alcaligenes Xylosoxidans, [[Alcaligenes Xylosoxidans]])
- Anaerobes: sometimes found in lung abscesses
- Escherichia Coli (see Escherichia Coli, [[Escherichia Coli]])
- Klebsiella (see Klebsiella, [[Klebsiella]]): increasing prevalence
- Pandoraea
- Proteus (see Proteus, [[Proteus]])
- Ralstonia
- Tuberculosis (see Tuberculosis, [[Tuberculosis]]): sporadic cases
Pulmonary Function Test (PFT’s) (see Pulmonary Function Tests, [[Pulmonary Function Tests]])
- Earliest finding is small airways disease
- Obstruction (typical): increased RV:TLC ratio, increased TLC, decreased DLCO, bronchodilator response (seen in 40% of cases, but some cases have paradoxical response to bronchodilators)
- Restriction may be superimposed late in course
- Exercise tolerance: related degree of obstruction
- Ventilatory muscle endurance: high (but without increased exercise tolerance)
- Weight training: one study showed 12% decrease in RV:TLC
- VO2 max: better predictor of survival than PFT’s
- Resting energy expenditure: increased (probably due to increased work of breathing)
CXR/Chest CT Patterns
- Hyperinflation: seen early increases with age
- Bronchiectasis: RUL usually most severely affected
- Mucous impaction: finger-like projections
- Subpleural cysts on mediastinal surface of upper lobes
- Prominent pulmonary arteries
Stool Pancreatic Elastase
- Useful to Diagnose Pancreatic Exocrine Insufficiency
- Has replaced prior use of measurement of fecal fat
Kidney/Ureter/Bladder (KUB) X-Ray
- Pancreatic calcifications may be seen (do not correlate with pancreatitis though)
Immunoglobulin Levels
- May be related to age or establishment of infection
- <10: decreased IgG (22%), increased IgG (7%)
- 10-20: increased IgG (25%)
- >20: increased IgG (47%)
Serum Immunoglobulin E (IgE)
- Elevated (22% of cases)
Serum Aspergillus Precipitins
- Positive (37% of cases)
Immediate Hypersensitivity to Aspergillus
- Present in 39%
Skin Testing
- Increased prevalence of atopy
CH50
- Normal
Alternate Complement
- Decreased (usually with severe lung disease)
Serum Immune Complexes
- Elevated (usually with severe lung disease)
Sweat Chloride Test
Technique
- Using Pilocarpine Iontophoresis: pilocarpine is a cholinergic agonist
- Checked on Two Different Occasions to Verify Result
Interpretation of Sweat Chloride Values
- Normal Sweat Chloride: <40 mEq/L
- Borderline Sweat Chloride: 40-60 mEq/L
- Abnormal Sweat Chloride: >60 mEq/L
False-Negative Sweat Chloride Test
- Point Mutation in Intron 19 of CFTR Gene
False-Positive Sweat Chloride Test
- Adrenal Insufficiency (see Adrenal Insufficiency, [[Adrenal Insufficiency]])
- Ectodermal Dysplasia
- Familial Cholestasis Syndrome
- Fucosidosis
- Glycogen Storage Disease Type 1
- Hypogammaglobulinemia
- Hypoparathyroidism (see Hypoparathyroidism, [[Hypoparathyroidism]])
- Hypothyroidism (see Hypothyroidism, [[Hypothyroidism]])
- Malnutrition
- Mauriac’s Syndrome
- Mucopolysaccharidosis
- Nephrogenic Diabetes Insipidus (see Diabetes Insipidus, [[Diabetes Insipidus]])
- Pancreatitis (see xxxx, [[xxxx]])
- Prostaglandin E1 Infusion (see Prostaglandin E1, [[Prostaglandin E1]])
- Pseudohypoaldosteronism
Genetic Analysis
- There are currently over 1900 mutations of the CFTR known
- Mutations in CFTR gene may be diagnostic in some cases where sweat chloride is normal (but not all mutations are known)
Screening
- Screening of Parents
- Detects 70% of carriers
- Screening of Newborns
- Performed in several states: however, number of mutations required to be screened to achieve approximate 99% sensitivity varies depending on diversity of population in the state
- Variable sensitivity from state to state (depending on the number of mutations being screened for): approximately 99%
Clinical Manifestations
General Comments
- Usually diagnosed in childhood
Dermatologic Manifestations
Sweat Gland Dysfunction
- Physiology: high loss of salt in sweat
- Clinical
- Increased Risk of Dehydration
- Salty Skin
Endocrinologic Manifestations
Diabetes Mellitus (DM) (see Diabetes Mellitus, [[Diabetes Mellitus]])
- Epidemiology
- Clinical
Gastrointestinal Manifestations
Biliary Cirrhosis (see xxxx, [[]])
- Epidemiology: occurs in 25% of cases
- Clinical
- Focal biliary cirrhosis cases usually manifest only elevated alkaline phosphatase and do not respond to dietary treatment
- However, symptomatic cases (2-5% of all cases) have typical findings of liver disease and respond to dietary treatment
Cholelithiasis
- Epidemiology: occurs in 12% of older cases
- Clinical: may present with biliary colic
Distal Intestinal (Partial or Complete) Obstruction (see Small Bowel Obstruction, [[Small Bowel Obstruction]])
- Epidemiology: occurs in 20% of cases
- Clinical:
- Usually occurs in ileum
- Appendiceal non-filling (without appendicitis) during contrast enema is common
Failure to Thrive
- Epidemiology: occurs in 10% of cases
Gastroesophageal Reflux Disease (GERD) (see Gastroesophageal Reflux Disease, [[Gastroesophageal Reflux Disease]])
- Epidemiology: occurs in 20% of cases
- Clinical: use of bethanecol may severely worsen lung function
Non-Alcoholic Fatty Liver Disease (NAFLD) (see Non-Alcoholic Fatty Liver Disease, [[Non-Alcoholic Fatty Liver Disease]])
- Epidemiology: fatty liver occurs in 30% of cases
- Physiology: due to malnutrition and other causes
Malabsorption-Related Diarrhea Due to Exocrine Pancreatic Insufficiency (see Diarrhea, [[Diarrhea]]): substantive pancreatic function remains in only 5-10% of cases
- Normal Carbohydrate Absorption
- Poor Nutritional Status
- Protein Malabsorption
- Steatorrhea
- Vitamin A/D/E/K Malabsorption
Meconium Ileus
- Epidemiology: occurs in 5-15% of cases
- Virtually diagnostic of CF
- May perforate
Rectal Prolapse
- Epidemiology
- Occurs in 20% of childhood CF cases, infrequent in adult CF cases
- CF is most common cause of rectal prolapse
- Physiology: prolapse occurs due to coughing, loss of perirectal fat, etc.
- Clinical
- May occur in association with pneumatosis coli
Recurrent Abdominal Pain Due to Duodenal Irritation (see Abdominal Pain, [[Abdominal Pain]])
- Physiology: due to poor buffering of gastric acid
Symptomatic Pancreatitis (see Acute Pancreatitis, [[Acute Pancreatitis]])
- Epidemiology: occurs in <1% of cases
- Clinical: usually occurs in cases with intact exocrine function
Weight Loss (see Weight Loss, [[Weight Loss]])
- Epidemiology
Genitourinary Manifestations
Delayed Puberty
- Epidemiology
Endocervicitis
- Epidemiology
Female Infertility
- Epidemiology: 20% of cases are infertile (due to anovulation, thick mucous, etc)
Male Infertility
- Epidemiology: 95% have abnormal Wolffian ducts, only 2-3% are fertile
Otolaryngologic Manifestations
Chronic Rhinitis (see Rhinitis, [[Rhinitis]])
- Epidemiology
Nasal Polyps (see Nasal Polyps, [[Nasal Polyps]])
- Epidemiology: occurs in 15-20% of cases
- Clinical
- Most common in teens
- May widen nose bridge
Recurrent Acute or Chronic Sinusitis (see Acute Rhinosinusitis, [[Acute Rhinosinusitis]])
- Diagnosis: typically diagnosed by CT
Pulmonary Manifestations
General Comments
- Generally, lung disease progresses more slowly in patients with intact exocrine pancreatic function, possibly due to avoidance of linoleic acid deficiency
Bronchiectasis (see Bronchiectasis, [[Bronchiectasis]])
- Epidemiology: usually present by teens or even earlier
- Clinical: usually found in upper>lower lobes
Bronchiolitis (see Bronchiolitis, [[Bronchiolitis]])
- Epidemiology: most common during the first 2 years
- Clinical: wheezing
Bronchiolitis Obliterans (BO) (see Bronchiolitis Obliterans, [[Bronchiolitis Obliterans]])
- Epidemiology: extent of BO is correlated with age of death
Chronic Cough (see Cough, [[Cough]])
- Epidemiology: usually an early manifestation
- Clinical: usually worse at night and in the early morning
Hemoptysis (see Hemoptysis, [[Hemoptysis]])
- Epidemiology: common in later stages of disease
- Physiology: due to enlarged bronchial arteries, bronchiectasis, infection
- Clinical
- Massive hemoptysis occurs in 5% of cases
- Most common source of massive hemoptysis is the right upper lobe
- Massive hemoptysis event does not alter the overall prognosis of CF
Lobar/Segmental Atelectasis(see Atelectasis, [[Atelectasis]])
- Epidemiology
- Occurs in only 5% of cases
- Most common in first 5 years of life, then decreases in frequency thereafter
- Clinical
- Usually R>L
- May be associated with mucous impaction in some cases
Pneumothorax (see Pneumothorax, [[Pneumothorax]])
- Epidemiology
- Sex: M=F
- Incidence of Pneumothorax Increases with Age: also notably, incidence has increased since the 1960’s
- Physiology: due to rupture of cysts
- Clinical
- R>L, but may be bilateral or tension
- Recurrence is common after first event
Pulmonary Hypertension/Cor Pulmonale (see Pulmonary Hypertension, [[Pulmonary Hypertension]])
- Epidemiology
- Occurs, on average, 8 months before death
- Physiology
- May be due to nocturnal desaturations
- Expected when pO2 is sustained <55 mm Hg
Recurrent Sinopulmonary Infection/Bronchitis/Pneumonia (see Pneumonia, [[Pneumonia]])
- Epidemiology
- May be exacerbated by smoke exposure
- Clinical
- Tenacious, purulent sputum
- Hyperinflation is usually present
- Wheezing: common in first 2 years
- Bronchial reactivity is unrelated to severity of lung disease or atopy (but is worse during winter months)
- CXR: usually peribronchial pattern
- Staphylococcus Aureus empyema has been described in infants, but pleural involvement is uncommon
Respiratory Failure/Chronic Hypoventilation (see Respiratory Failure, [[Respiratory Failure]]) and Chronic Hypoventilation (see Chronic Hypoventilation, [[Chronic Hypoventilation]])
- Clinical: xxx
Thick, Tenacious Mucus
- Physiology: due to inadequate hydration of the airway lining fluid secondary to impaired chloride movement
- Clinical: xxx
Renal Manifestations
Metabolic Alkalosis (see Metabolic Alkalosis, [[Metabolic Alkalosis]])
- Epidemiology: occurs in in young childhood cases but rare in older children and adults
- Clinical
- Hypochloremia
- Hyponatremia (see Hyponatremia, [[Hyponatremia]])
Rheumatologic Manifestations
Clubbing (see Clubbing, [[Clubbing]])
- Epidemiology
- Occurs in almost all cases
- Seen early in course
- Clinical
- Severity of clubbing correlates with degree of lung disease
Hypertrophic Pulmonary Osteoarthropathy (HPO) (see xxxx, [[xxxx]])
- Epidemiology: occurs in 15% of older adolescent and adults cases
- Clinical
- Most common in tib-fib, radius, and ulna
- HPO correlates with degree of lung disease
Small Stature
- xxx
Other Manifestations
- xxx
Treatment
Antibiotics
Pseudomonas Aeruginosa (see Pseudomonas Aeruginosa, [[Pseudomonas Aeruginosa]])
- Treat organisms recovered early (better than delaying)/ treat with higher doses (to get levels in lung secretions)/treat for longer (up to 3-4 weeks)
- PO: various (ciprofloxacin decreases Pseudomonas in CF but resistance develops rapidly)
- IV: double coverage
- Nebulized (Tobramycin): decrease hospitalizations/resistance is not a problem
Staphylococcus Aureus (see Staphylococcus Aureus, [[Staphylococcus Aureus]])
- Studies in young children with CF in previously good health who received PO cephalosporin had decreased Staph aureus colonization rates but had higher rate ofPseudomonas aeruginosa colonization (this colonization was also associated with increased pulmonary symptoms)
- Treatment of Staphylococcus Aureus had no impact on hospitalizations, lung function, or respiratory symptoms
CFTR-Active Agents
Ivacaftor (Kalydeco) (see Ivacaftor, [[Ivacaftor]])
- Indications
- Cystic Fibrosis with G551D Mutation in At Least One of Their CFTR Genes: FDA-Approved for this Indication in 2012
- General Information
- G551D Mutation Occurs in Only 4.4% of CF Patients
- G551D is a “Gating Mutation”: this mutation impairs the regulated opening of the ion channel that is formed by the CFTR protein
- Ivacaftor is Indicated for CF Patients >2 y/o
- General Information
- Cystic Fibrosis with Other CFTR “Gating Mutations”: G1244E, G1349D, G178R, G551S, R117H, S1251N, S1255P, S549N, S549R -> FDA-approved for these mutants
- Cystic Fibrosis with G551D Mutation in At Least One of Their CFTR Genes: FDA-Approved for this Indication in 2012
- Pharmacology: ivacaftor restores function of the mutant ion channel in CF patients with “gating mutations”
- Clinical Efficacy
- VX08-770-102 Study Group (NEJM, 2011) [MEDLINE]
- Ivacaftor improved lung function (FEV1) at 2 wks, which was sustained through 48 wks
- Ivacaftor improved pulmonary exacerbations: 55% risk reduction
- Ivacaftor improved patient-reported respiratory symptoms, weight, and sweat chloride concentrations
- Ivacaftor decreased serious adverse events
- Longitudinal Cohort Study of Ivacaftor in CF with G551D Mutation (Am J Resp Crit Care Med, 2014) [MEDLINE]
- Ivacaftor improved FEV1 and BMI at 6 mo
- Ivacaftor decreased hospitalization rate at 6 mo
- Ivacaftor decreased Pseudomonas Aeruginosa burden at 6 mo
- UK/Irish Study of Effects of Ivacaftor in CF with Severe Lung Disease and G551D Mutation (Chest, 2014) [MEDLINE]: follow-up data for median of 237 days (range: 125-270 days)
- Ivacaftor improved FEV1 (significant effect noted after 90 days of treatment)
- Ivacaftor improved weight
- Ivacaftor improved number of inpatient intravenous antibiotic days
- Study of Ivacaftor in a CF Patient with a Non-Gating (Compound Heterozygous P67L/F508del CFTR Genotype) Mutation (Chest 2015) [MEDLINE]: ivacaftor x 1 yr resulted in clinical improvement, normalization of spirometry, and dramatic reduction in radiographic structural airway changes
- VX08-770-102 Study Group (NEJM, 2011) [MEDLINE]
Lumacaftor + Ivacaftor (Orkambi) (see Lumacaftor + Ivacaftor, [[Lumacaftor + Ivacaftor]])
- Indications
- Cystic Fibrosis with Homozygous F508del (Phe508del) Mutation (FDA-Approved for this Indication in 7/15)
- General Information
- F508del is present in 68% of CF Cases
- The F508del Mutation Interferes with CFTR Protein Folding and Channel Gating Activity
- Lumacaftor + Ivacaftor is Indicated for Patients at Least 12 y/o
- General Information
- Cystic Fibrosis with Homozygous F508del (Phe508del) Mutation (FDA-Approved for this Indication in 7/15)
- Pharmacology: lumacaftor partially corrects CFTR protein folding, while ivacaftor improves CFTR gating activity
- Neither Drug (Lumacaftor or Ivacaftor) is Efficacious When Used Alone in CF Patients with the F508del Mutation
- Clinical Efficacy
- TRAFFIC and TRANSPORT Trials (NEJM, 2015) [MEDLINE]
- Lumacaftor + Ivacaftor Improved FEV1
- Lumacaftor + Ivacaftor Decreased Pulmonary Exacerbations
- Lumacaftor + Ivacaftor Decreased Antibiotic Use and Rate of Hospitalizations
- TRAFFIC and TRANSPORT Trials (NEJM, 2015) [MEDLINE]
Intravenous Immunoglobulin (IVIG) (see Intravenous Immunoglobulin, [[Intravenous Immunoglobulin]])
- Effective (with Intravenous Antibiotics: as assessed by PFT’s
Bronchoscopy (see Bronchoscopy, [[Bronchoscopy]])
- No Clinical Benefit
Postural Drainage with Chest Physiotherapy (1-4x/day)
- Has been shown to increase secretion clearance (as assessed by radioactive tracer techniques)
- The effect on FEV1, frequency of exacerbations, quality of life and mortality have not been substantiated in adequate studies
Aerosolized Saline
- No Clinical Benefit
Aerosolized Hypertonic Saline (see Hypertonic Saline, [[Hypertonic Saline]])
- xxxx
Bronchodilators
- Useful for bronchospasm, but long-term benefit unknown (animal studies show hypersecretory state with long-term use of ß-agonists)
- Chromolyn is not effective
- Theophylline is used, but may be poorly tolerated (GI side effects)
Oxygen (see Oxygen, [[Oxygen]])
- Not proven to prevent onset of cor pulmonale (as it does in chronic bronchitis)
Corticosteroids (see Corticosteroids, [[Corticosteroids]])
- Useful only for broncho-spasm or ABPA, as studies do not show benefit
Non-Steroidal Anti-Inflammatory Drugs (NSAID) (see Non-Steroidal Anti-Inflammatory Drug, [[Non-Steroidal Anti-Inflammatory Drug]])
- Advocated for airway inflammation but unproven
Cough Suppressants
- Avoid use
Iodides
- May cause goiter or hypothyroidism long-term in CF
Mucolytics
- N-Acetylcysteine (Mucomyst) (see N-Acetylcysteine, [[N-Acetylcysteine]]: may be useful short-term but is irritating long-term
Dornase Alfa (Pulmozyme) (see Dornase Alfa, [[Dornase Alfa]])
- Pharmacology: DNase
- Clinical Efficacy: studies show 5.6-5.8% increase in FEV1, 3.0-3.8% decrease in FVC, decreased risk of exacerbations to 37%, fewer hospital days, better well-being, fewer IV antbx days
- However, cost effectiveness is unknown
Aerosolized Elastase Inhibitors
- xxx
Amiloride (see Amiloride, [[Amiloride]])
- Pharmacology: may improve mucous clearance
ATP (and UTP)
- Pharmacology: nasal mucosal secretagogues in CF (increase intracellular calcium concentration)
Exercise
- Physiology
- Increase ventilatory muscle endurance (and exercise tolerance) but do not affect PFT’s
- May strengthen cough
Gene Therapy
- Pharmacology: with treatment, respiratory epithelium expresses CFTR for days-wks
Nutrition/Pancreatic Enzyme/Vitamins
- Increased work of breathing results in increased caloric need -> supports use of aggressive nutrition regimen
- Tube feeds increase weight but do not affect PFT’s
- Better nutrition and intact exocrine function are associated with slower lung disease progression
Treatment of Pneumothorax (see Pneumothorax, [[Pneumothorax]])
- Thoracoscopy or Pleurodesis: due to high rate of recurrence
Treatment of Gastrointestinal Complications
- Meconium ileus, intestinal obstruction treated by Gastrograffin enema (or intestinal flush with NG saline) or surgery
- Rectal prolapse can be reduced voluntarily
- Ursodeoxycholic acid improves LFT’s
- Treatment of Liver Disease: standard treatment
- Liver Transplant (see Liver Transplant, [[Liver Transplant]]): liver transplant in CF has >50% 2 year survival
Treatment of Nasal Polyps (see Nasal Polyps, [[Nasal Polyps]])
- Polypectomy
Prevention
- Influenza Vaccination (see Influenza Virus, [[Influenza Virus]]): indicated
- Measles (Rubeola) Vaccination (see Measles Virus, [[Measles Virus]]): indicated
- Pertussis Vaccination (see Pertussis, [[Pertussis]]): indicated
- Pneumococcal Vaccination (see Streptococcus Pneumoniae, [[Streptococcus Pneumoniae]]): not shown to be efficacious
- German Measles (Rubella) Vaccination (see Rubella Virus, [[Rubella Virus]]): indicated
Lung Resection
- Indication
- Hemoptysis (see Hemoptysis, [[Hemoptysis]])
- Recurrent Infection
Lung Transplantation (see Lung Transplant, [[Lung Transplant]])
- Indications
- FEV1 <30% Predicted: earlier for female or younger cases
- Prognosis: the 2-year survival for double-lung transplant is >50%
Prognosis
Median Survival Rates
- Historical Median Survival Rates
- Median Survival in 1960: 5 years
- Median Survival in 1970: 19 years
- Projected Median Survival for Patients with CF Born in 2010 [MEDLINE]
- Male: 40 years (CI, 39 to 42 years)
- Projected to be >50 years if CF mortality continues to decrease at the rate observed between 2000 and 2010
- Female: 37 years (CI, 35 to 39 years)
- Male: 40 years (CI, 39 to 42 years)
Risk of Death is Predicted by FEV1
- Relative Risk of Death Within 2 yrs is 2x for each 10% FEV1 Below Predicted [MEDLINE]
Positive Survival Predictors
- Black Race
- Care in Specialized Center
- Male Sex
- Northern Climate
Negative Survival Predictors
- Colonization with Pseudomonas (see Pseudomonas, [[Pseudomonas]])
- Delta F508 Mutation [MEDLINE]
- Female Sex [MEDLINE]
- Massive Hemoptysis (see Hemoptysis, [[Hemoptysis]])
- Pneumothorax (see Pneumothorax, [[Pneumothorax]])
- Hypercapnia (pCO2 >50 mm Hg) + Hypoxemia (pO <55 mmg Hg) + FEV1 <30% Predicted: <50% of patients survive for >24 mo
References
General
- xxx
Mortality Rates
- Prediction of mortality in patients with cystic fibrosis. N Engl J Med. 1992 Apr 30;326(18):1187-91 [MEDLINE]
- Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J 2007;29:522-526 [MEDLINE]
- Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation Patient Registry. Ann Intern Med 2014;161:233-241 [MEDLINE]
Treatment
- Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 2010;363(21):1991-2003 [MEDLINE]
- A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365(18):1663-1672 [MEDLINE]
- Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med. 2013 Jun;187(11):1219-25 [MEDLINE]
- Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp-CFTR mutation: a phase 3, open-label extension study (PERSIST). Lancet Respir Med. 2014 Nov;2(11):902-10. Epub 2014 Oct 9 [MEDLINE]
- Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am J Respir Crit Care Med. 2014 Jul 15;190(2):175-84. doi: 10.1164/rccm.201404-0703OC [MEDLINE]
- Effects of ivacaftor in cystic fibrosis patients carrying the G551D mutation and have severe lung disease. Chest 2014;146(1):152-158 [MEDLINE]
- A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med. 2014 Jul;2(7):527-38. doi: 10.1016/S2213-2600(14)70132-8. Epub 2014 Jun 24 [MEDLINE]
- Improved clinical and radiographic outcomes after treatment with ivacaftor in a young adult with cystic fibrosis with the P67L CFTR mutation. Chest. 2015 Mar;147(3):e79-82. doi: 10.1378/chest.14-1198 [MEDLINE]