Scleroderma (see Scleroderma): antibodies against VWF
Systemic Lupus Erythematosus (see Systemic Lupus Erythematosus): antibodies against VWF (higher MW multimers appear to be more susceptible to degradation by this mechanism)
Dextran (see Dextran): branched polysaccharides of 40 or 70 kDa which inhibit platelet factor-3 availability, adsorb to platelets, and decrease plasma factor VIII-vWF -> inhibit platelet aggregation + secretion
Aortic Stenosis (see Aortic Stenosis): high intravascular shear forces -> increased clearance of high MW multimers (possibly due to increased susceptibility to serum proteases)
Disseminated Intravascular Coagulation (DIC) (see Disseminated Intravascular Coagulation): hyperfibrinolytic state -> VWF degradation by proteolytic enzymes (such as plasmin)
Essential Thrombocythemia (see Essential Thrombocythemia): increased numbers of platelets may result in increased VWF binding -> increased VWF clearance
Lymphoma (see Lymphoma): adsorption of VWF to lymphoma cells
Heyde’s Syndrome = constellation of aortic stenosis + gastrointestinal angiodysplasia -> high intravascular shear forces across the stenotic aortic valve increase clearance of high MW multimers
Hemoglobinopathies
Hypothyroidism (see Hypothyroidism): decreased synthesis of VWF
Post-Multiple Transfusions: due to development of antibodies against VWF
Bernard-Soulier Syndrome (see Bernard-Soulier Syndrome): autosomal recessive deficiency in platelet glycoprotein Ib-IX complex -> inability to bind Von Willebrand factor
Dextran (see Dextran): branched polysaccharides of 40 or 70 kDa which inhibit platelet factor-3 availability, adsorb to platelets, and decrease plasma factor VIII-VWF -> inhibit platelet aggregation + secretion
Glanzmann’s Thrombasthenia (see Glanzmann’s Thrombasthenia,): autosomal recessive deficiency or defect in IIb-IIIa complex -> platelets cannot bind fibrinogen -> inability of platelets to aggregate
Phsyiology: adenosine triphosphate depletion results in decreased platelet aggregation
Omega-3 Fatty Acids/Fish Oil (see Omega-3 Fatty Acid): increase PGI3 synthesis (a more potent platelet inhibitor than prostacyclin, PGI2), increase thromboxane A3 (a less potent platelet activator than thromboxane A2)
Diets naturally rich in omega 3 fatty acids can result in a prolonged bleeding time and abnormal platelet aggregation studies -> the actual associated bleeding risk is unclear
Vitamin E (see Vitamin E): inhibits protein kinase C–mediated platelet aggregation and nitric oxide synthesis
Platelet Secretion Defect
Cyclooxygenase Defect
Inherited Platelet Secretion Defect: defect of cyclooxygenase-1 activity
Cyclooxygenase (COX) Inhibitors
Aspirin (see Acetylsalicylic Acid): inrreversible inhibition of cyclooxygenase-1 activity -> inhibited synthesis of TXA2 (an important mediator of platelet secretion and aggregation)
Single dose of aspirin can impair hemostasis for 5-7 days
Effect on platelet function (as assessed by aggregometry) can persist for up to 7 days (although it has frequently returned to normal by 3 days after the last dose)
Other Non-Selective COX-1/COX-2 Inhibitor NSAID’s (see Nonsteroidal Anti-Inflammatory Drug): however, these agents have reversible and less sustained COX inhibition (and anti-platelet effects) than aspirin
Note: the selective COX-2 inihibitors, celecoxib (Celebrex) and rofecoxib (Vioxx), do not have anti-platelet effects
Dextran (see Dextran): branched polysaccharides of 40 or 70 kDa which inhibit platelet factor-3 availability, adsorb to platelets, and decrease plasma factor VIII-vWF -> inhibit platelet aggregation + secretion
Cardiopulmonary Bypass (see Cardiopulmonary Bypass): platelet activation in bypass circuit (due to artificial membrane) -> prolonged bypass may result in platelet degranulation with a transient platelet storage pool disorder
Treatment: platelet transfusion
Cirrhosis/End-Stage Liver Disease (ESLD) (see Cirrhosis): some patients manifest permanent platelet degranulation
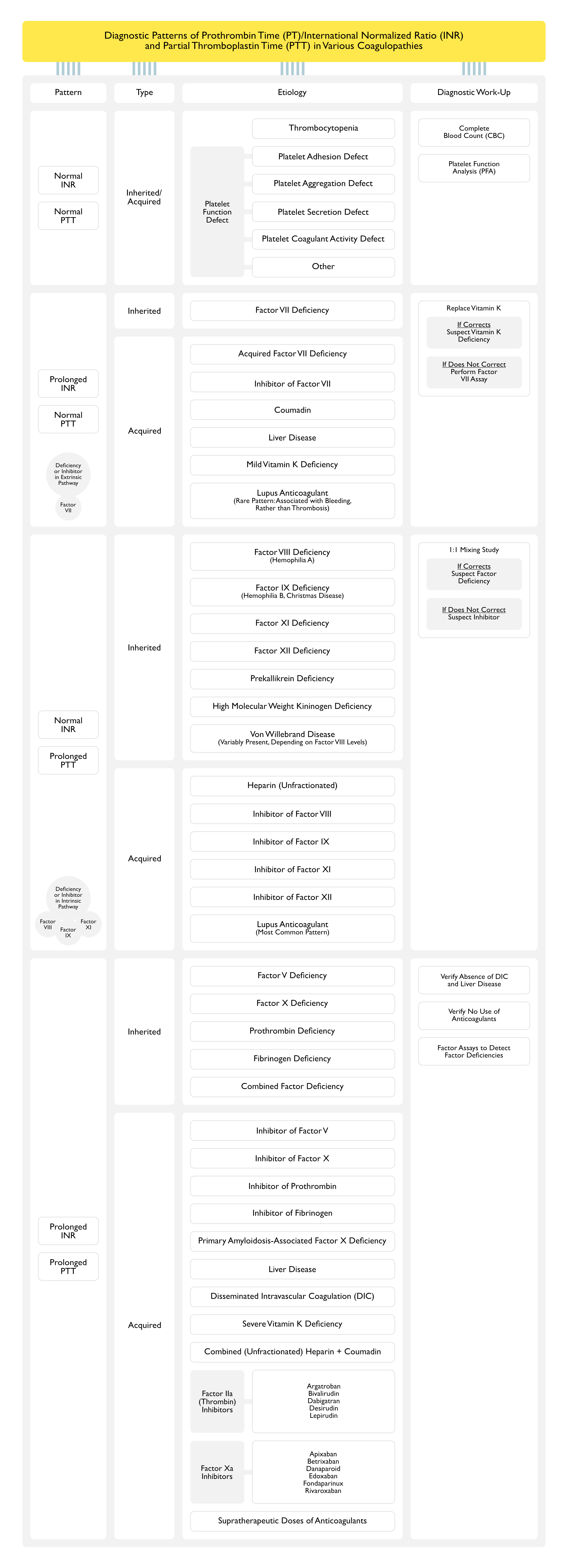
Deficiency/Inhibitors of factors VIII, IX, XI, XII, as well as common pathway factors
However, PTT is less sensitive for deficiency/inhbitors of common pathway factors -> this is why vitamin K deficiency (with decreased factors II, VII, IX, X) prolongs INR more than PTT
Rationale: measures how fast given amount of thrombin turns fibrinogen into fibrin -> therefore, is designed to detect quantitative or qualitative problems with fibrin
Can be prolonged by high levels of fibrin degradation products
Very sensitive to heparin
Reptilase Time
Rationale: used to determine if prolonged thrombin time is due to heparin
Insensitive to Heparin
1:1 Mix
Rationale:
Technique: mix control plasma (with 100% activity of each factor) + patient plasma to determine if a factor deficiency vs inhibitor is present
As factor levels of 50% or higher are sufficient for a normal INR/PTT, 1:1 mix will correct INR/PTT in presence of a factor deficiences, but not in presence of a factor inhibitor
PTT with Polybrene or Heparinase
Rationale: used to neutralize heparin in the sample to determine if prolonged PTT is due to heparin
PTT with Excess Phospholipid
Rationale: used to detect presence of anti-phospholipid antibody (lupus anticoagulant, etc)
Rationale: assesses for platelets defects (relatively insensitive for factor deficiencies or inhibitors)
Platelet Function Analysis (PFA)
Rationale: Essentially an in vitro bleeding time assay which assesses different components of platelet activation (in response to ADP, epinephrine, collagen)
Von Willebrand Factor Antigen
Rationale: quantifies the amount of Von Willebrand factor -> decreased in Von Willebrand Disease
Rationale: evaluates function of Von Willebrand factor by testing ristocetin-induced platelet aggregation of normal platelets in presence of patient’s plasma
Most sensitive and specific test for Von Willebrand disease -> decreased in all types of Von Willebrand disease
Assesses the binding of Von Willebrand factor to platelet GP1b
Evaluates function of Von Willebrand factor by testing ristocetin-induced platelet aggregation of patient’s platelets in presence of patient’s plasma
Less sensitive and specific than ristocetin cofactor test
Usually decreased in Von Willebrand disease, but may be normal in some cases
In type IIB Von Willebrand disease: platelets are hyperresponive to ristocetin (platelets aggregate in response to abnormally low ristocetin concentration)
Factor VIII Activity/Level
Factor VIII activity assay is performed -> level is inferred from activity: decreased in factor VIII deficiency and Von Willebrand disease
Von Willebrand factor normally carries factor VIII (this prolongs factor VIII half-life) -> therefore, if Von Willebrand factor binding of factor VIII is impaired or Von Willebrand factor is low, then, factor VIII level is low
Diagnostic Patterns of Prothrombin Time (PT)/International Normalized Ratio (INR) and Partial Thromboplastin Time (PTT) in Various Coagulopathies
Stepwise Evaluation of Coagulopathy
Step 1 (assessment for platelet problem): platelet count, PFA
Step 2 (assessment for single factor deficiency): INR/PTT, factor assays
Step 3 (assessment for multiple factor deficiency): INR/PTT, thrombin time, factor assays
Step 4 (assessment for circulating anticoagulant): PTT with polybrene or heparinase, PTT with 1:1 mix, PTT with excess phospholipid, thrombin time
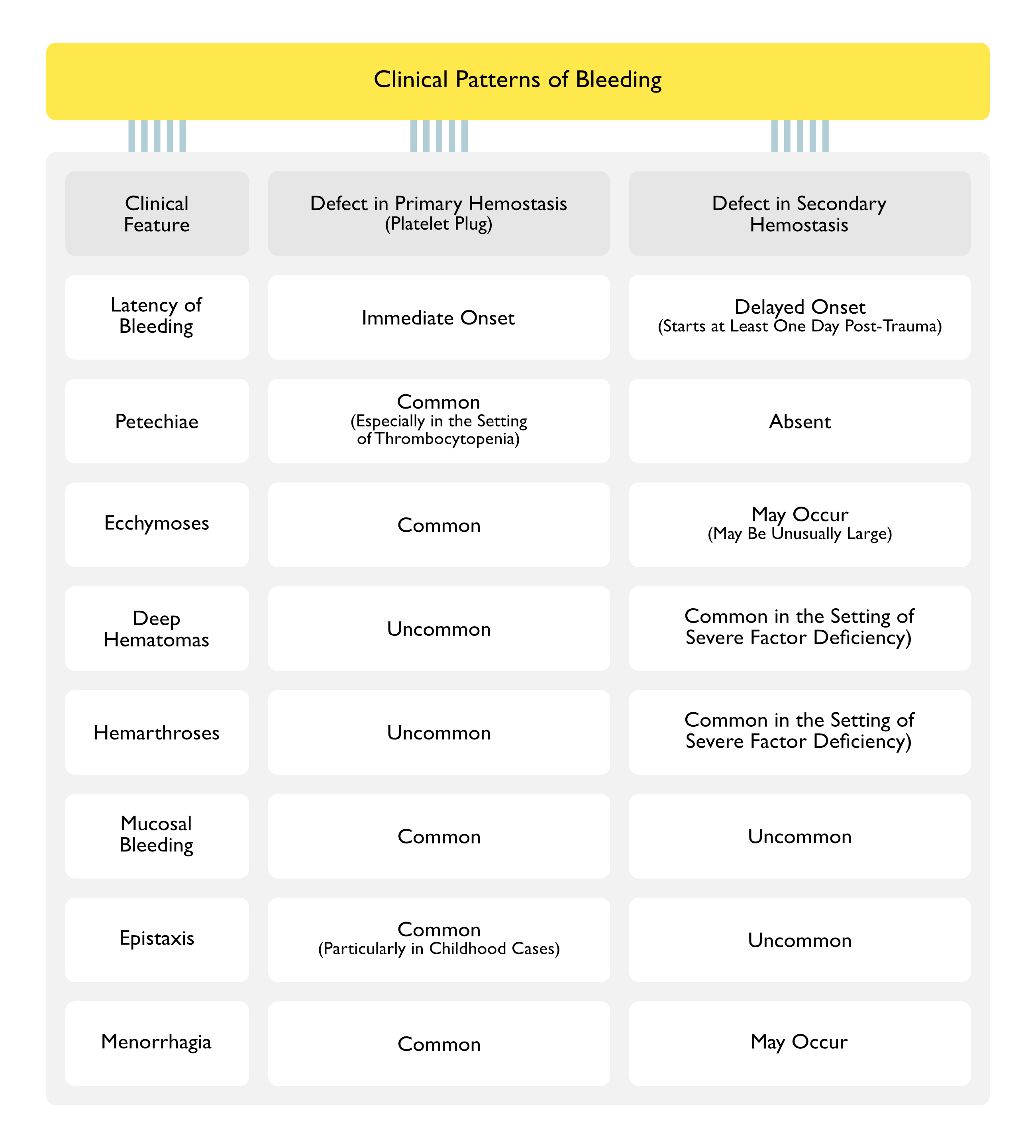
Clinical
Locations of Hemorrhage
Central Nervous System Hemorrhage: central nervous system hemorrhage is the major cause of bleeding-related deaths in patients with severe congenital factor deficiencies
Von Willebrand Disease (see Von Willebrand Disease): epistaxis is a common symptom in young males
Excessive Menstrual Bleeding
Menorrhagia (see Menorrhagia): loss of >80 mL of blood per cycle (or >4 super pads or tampons per day) or menses lasting >7 days
Gastrointestinal Hemorrhage (see Gastrointestinal Hemorrhage): gastrointestinal hemorrhage in presence of a bleeding disorder is usually associated with underlying gastrointestinal tract pathology
Von Willebrand Disease (Especially Types 2 and 3): has been associated with angiodysplasia of the bowel and gastrointestinal hemorrhage
Common in women with underlying bleeding disorders
In women with type 1 Von Willebrand Disease and symptomatic hemophilia carriers in whom levels of Von Willebrand factor and factor VIII usually normalize during pregnancy, the onset of post-partum hemorrhage may be delayed
Women with a history of postpartum hemorrhage have a high risk of recurrence with subsequent pregnancies
Post-Colonoscopic Polypectomy (see Colonoscopy): delayed bleeding may occur
Post-Tonsillectomy (see Tonsillectomy): Bleeding may occur early after surgery or after approximately 7 days postoperatively (with loss of the eschar at the surgical site)