Incidence: 3 cases/1 million adults (Oklahoma TTP-HUS Registry Data) [MEDLINE]
Median Age at Time of Diagnosis: 41 y/o (range: 9-78 y/o)
Very Rare in Children: 30x less common than in adults
Risk Factors
Black Race: blacks (Oklahoma TTP-HUS Registry Data) [MEDLINE]: 36% of cases were black
Female Sex (Oklahoma TTP-HUS Registry Data) [MEDLINE]: 76% of cases were female
Physiology
Background-Von Willebrand Factor (vWF)
Von Willebrand Factor (vWF) is a Long Stringlike Molecule Synthesized by Vascular Endothelial Cells and Megakaryocytes
vWF is Synthesized as a Single VWF Precursor: cleavage and assembly into disulfide-linked multimers occurs in plasma
VWF Activity is Distributed Among a Series of Plasma Multimers: ranging in size from MW 400k-20 million
Modest Reduction in Plasma VWF Concentration OR Selective Loss of One of the High Molecular Weight Multimers Results in Decreased Platelet Adhesion and Clinical Bleeding
Locations of Von Willebrand Factor
VWF Circulates as a Series of Multimers Formed from a Basic Dimer Subunit
Ultralarge Von Willebrand Factor Multimers Attach to the Endothelial Surface
Von Willebrand Factor Binds to the Platelet GP1b Receptor
ADAMTS13 (A Disintegrin And Metalloprotease with a Thrombospondin Type 1 Motif, Member 13) is a Von Willebrand Factor-Cleaving Plasma Protease
Cleavage by ADAMTS13 is a Normal Function Which Prevents Ultralarge Von Willebrand Factor Multimers from Accumulating, Especially in Areas of High Shear Stress (Such as Small Arterioles and Capillaries)
Shear Stress Results in a Conformational Change in Von Willebrand Factor Multimers, Exposing the ADAMTS13 Cleavage Site
Physiologic Sites of ADAMTS13 Synthesis
Hepatic Stellate Cells: primary site of synthesis
Endothelial Cells: lesser site of synthesis
Megakaryocytes: lesser site of synthesis
Acquired Thrombotic Thrombocytopenic Purpura is a Thrombotic Microangiopathy Caused by Decreased Activity of ADAMTS13 (Due to an Acquired Autoantibody Inhibitor of ADAMTS13) (see Thrombotic Microangiopathy, [[Thrombotic Microangiopathy]])
Decreased ADAMTS13 Levels (<10% Activity) Result in Ultralarge Von Willebrand Factor Multimer Accumulation on the Endothelial Surface, Where Platelets Then Subsequently Accumulate
Severe Reduction in ADAMTS13 Activity is Required for Clinical Manifestations of TTP
Lesser decreases in ADAMTS13 may be observed in sepsis, liver disease, cardiac surgery, pancreatitis, and last two trimesters of pregnancy (lowest levels occur at 36-40 weeks of gestation and during early puerperium): these conditions are extremely unlikely to cause disease by themselves
However, Reduction in ADAMTS13 Activity Alone is Not Sufficient to Cause Disease: other factors such as acute inflammation or a prothrombotic stimulus may be required to trigger the onset of disease
In addition, plasmin may substitute for ADAMTS13 in cleaving ultralarge Von Willebrand factor multimers
Prothrombin Time (PT)/International Normalized Ratio (INR) and Partial Thromboplastin Time (PTT) (see Prothrombin Time, [[Prothrombin Time]] and Partial Thromboplastin Time, [[Partial Thromboplastin Time]])
Required to Exclude Disseminated Intravascular Coagulation (DIC)
Reticulocytes are Newly-Released RBC’s: they are slightly larger than mature RBC’s and have some residual ribosomal RNA -> presence of RNA allows for staining, with detection and counting
Normal Reticulocyte Percentage: 1-2%
Reticulocyte Production Index (RPI) = Reticulocyte Percentage x (Patient’s Hct/Normal Hct) x (1/RMT): corrects reticulocyte percentage for the degree of anemia (normalized to Hct 45%) and reticulocyte maturation time (RMT)
Use Normal Hct = 45%
RMT
Hct 45% -> RMT = 1.0 days
Hct 15% -> RMT = 2.5 days
Reticulocyte Production Index > or = to 2.5%: indicates adequate bone marrow response to anemia [MEDLINE]
Acute/Subacute Hemorrhage: note that acute hemorrhage may not result in an increased RPI, due to the time that it takes to increase epo synthesis and increase bone marrow RBC production
Hemolytic Anemia
Reticulocyte Production Index <2.5%: indicates inadequate bone marrow response to anemia [MEDLINE]
Chronic Anemia
Hypoproliferative Anemia: such as iron deficiency, marrow hyporesponsiveness, aplasia, etc
Maturation Disorder: such as vitamin B2 deficiency, etc
Reticulocytosis: common
Direct Coombs Test (Direct Anti-Globulin Test) (see Direct Coombs Test, [[Direct Coombs Test]])
Negative: due to absence of immune hemolysis
Indirect Coombs Test (Indirect Anti-Globulin Test)
Severely Decreased ADAMTS13 Activity (<10%): characteristic during an acute flare of the disease
However, if Disease is Suspected and ADAMTS13 Test Results are Not Available, Therapy Should Not be Delayed
Relationship of Disease to ADAMTS13 Activity
Severe Reduction in ADAMTS13 Activity is Required for Clinical Manifestations of TTP
However, Reduction in ADAMTS13 Activity Alone is Not Sufficient to Cause Disease: other factors such as acute inflammation or a prothrombotic stimulus may be required to trigger the onset of disease
Range of ADAMTS13 Levels in TTP in Harvard Thrombotic Microangiopathy Research Collaborative [MEDLINE]: bimodal distribution of ADAMTS13 activity was observed in suspected TTP cases
ADAMTS13 Activity <10%: found in 27% of cases -> this cohort was less likely to have an alternative explanation for MAHA and thrombocytopenia
ADAMTS13 Activity 56% (Range: 42-68%): found in 73% of cases -> this cohort was likely to have an alternative explanation for MAHA and thrombocytopenia
Decreased ADAMTS13 Activity Levels May Persist (Due to Presence of an Anti-ADAMTS13 Inhibitor Antibody) for Months-Years in Some Cases After Recovery: despite a lack of clinical symptoms
However, most cases demonstrate a recovery in ADAMTS13 activity levels with clinical recovery
Low ADAMTS13 Activity (10-50%): may be seen in hospitalized patients with sepsis/malignancies
In addition, activity levels of 10-20% may be seen in TTP patients who have received blood products (since blood products contain functional ADAMTS13)
Normal ADAMTS13 Activity (>50%): considered normal
Anti-ADAMTS13 Antibodies
May Be Detected: laboratories usually run this testing reflexively when ADAMTS13 activity levels are low
Tissue Biopsy
Pathologic Changes: small arteriolar/capillary platelet microthrombi with hyaline changes in/around the vessel walls
Changes are consistent with thrombotic microangiopathy -> there are no specific findings which TTP this from other thrombotic microangiopathies
Genetic Testing for ADAMTS13 Gene Mutations
May Be Used for Suspected Cases of Hereditary Thrombotic Thrombocytopenic Purpura (TTP) (see Thrombotic Thrombocytopenic Purpura-Hereditary, [[Thrombotic Thrombocytopenic Purpura-Hereditary]])
Testing Availability: through the Hereditary TTP (Upshaw-Schulman Syndrome) Registry (www.ttpregistry.net)
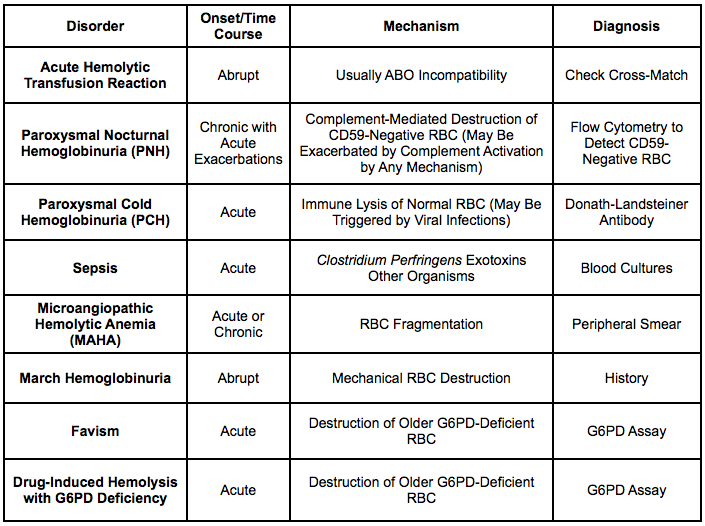
Clinical Differentiation of Hemolytic Syndromes
Clinical Differentiation of Intravascular Hemolysis Syndromes
Clinical Manifestations
General Comments
Course of Disease: may span days-weeks (but may continue for months in some cases)
Classic Pentad of Microangiopathic Hemolytic Anemia + Thrombocytopenia + Fever + Acute Kidney Injury + Neurologic Findings: rare (occurs in only 5% of cases)
Possibly due to presumptions that pulmonary microvasculature may be inherently resistant to the formation of platelet thrombi and pulmonary function can be maintained in spite of multiple microvascular thrombi
Thyroid Involvement: may occur
Cardiovascular Manifestations
Chest Pain with Elevated Serum Troponin (see Chest Pain, [[Chest Pain]] and Serum Troponin, [[Serum Troponin]])
Epidemiology: chest pain occurs in 22% of cases
However, Clinically-Significant Acute Myocardial Infarction/Arrhythmia is Rare
Other Less Common Cardiovascular Manifestations
Acute Myocardial Infarction (MI) (see xxxx, [[xxxx]])
Note: the timing of diarrhea is crucial, as diarrhea that precedes the onset of symptoms by several days may instead be indicative of Shiga toxin-producing Escherichia Coli hemolytic-uremic syndrome (see Shiga Toxin-Producing Escherichia Coli Hemolytic-Uremic Syndrome, [[Shiga Toxin-Producing Escherichia Coli Hemolytic-Uremic Syndrome]])
Fever/Chills (see Fever, [[Fever]]): occurs in <33% of cases (Blood, 2010) [MEDLINE]
Treatment
Plasma Exchange (see Plasmapheresis, [[Plasmapheresis]])
Required Urgently
Although fresh frozen plasma (FFP) infusion may be used as a temporizing measure, it is not a substitute for plasma exchange since it does not remove the ADAMTS13 inhibitor autoantibody and the volume of plasma that can be infused is far less than with plasma exchange (see Fresh Frozen Plasma, [[Fresh Frozen Plasma]]) [MEDLINE]
Prednisone or Methylprednisolone (Solumedrol) (see Prednisone, [[Prednisone]] or Methylprednisolone, [[Methylprednisolone]]))
Less Severe Disease with No Neurologic Abnormalities: prednisone 1 mg/kg/day PO
More Severe Disease: methylprednisolone 125 mg q6-12hrs IV
Rituximab (Rituxan) (see Rituximab, [[Rituximab]])
Indications
Refractory Disease
Investigational Therapies
Anti-VWF (Caplacizumab)
Recombinant ADAMTS13
References
Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393 [MEDLINE]
Late relapses in patients successfully treated for thrombotic thrombocytopenic purpura. Canadian Apheresis Group. Ann Intern Med. 1995 Apr 15;122(8):569-72 [MEDLINE]
Cellular source of serum lactate dehydrogenase elevation in patients with thrombotic thrombocytopenic purpura. J Clin Apher. 1998;13(1):16 [MEDLINE]
Thrombotic microangiopathy associated with reactivation of human herpesvirus-6 following high-dose chemotherapy with autologous bone marrow transplantation in young children. Bone Marrow Transplant 1999; 254: 919–923 [MEDLINE]
Hemolytic uremic syndrome in a child with leukemia and cytomegalovirus infection. Pediatr Nephrol 2000; 14: 1118–1120 [MEDLINE]
Specific von Willebrand factor-cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood 2001; 98: 1765–1771 [MEDLINE]
A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int. 2006 Aug;70(3):423-31 [MEDLINE]
Current management of thrombotic thrombocytopenic purpura. Curr Opin Hematol. 2008 Sep;15(5):445-50 [MEDLINE]
Brain lesions are most often reversible in acute thrombotic thrombocytopenic purpura. Neurology. 2009;73(1):66 [MEDLINE]
How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood. 2010;116(20):4060 [MEDLINE]
Children and adults with thrombotic thrombocytopenic purpura associated with severe, acquired Adamts13 deficiency: comparison of incidence, demographic and clinical features. Pediatr Blood Cancer. 2013 Oct;60(10):1676-82. Epub 2013 Jun 1 [MEDLINE]
Drug-induced thrombotic microangiopathy: a systematic review of published reports. Blood. 2015 Jan 22;125(4):616-8. doi: 10.1182/blood-2014-11-611335. Epub 2014 Nov 20 [MEDLINE]
Syndromes of thrombotic microangiopathy. N Engl J Med. 2014 Aug 14;371(7):654-66. doi: 10.1056/NEJMra1312353 [MEDLINE]
Pulmonary involvement in patients with thrombotic thrombocytopenic purpura. Eur J Haematol. 2014 Feb;92(2):156-63. Epub 2013 Nov 26 [MEDLINE]
Impact of severe ADAMTS13 deficiency on clinical presentation and outcomes in patients with thrombotic microangiopathies: the experience of the Harvard TMA Research Collaborative. Br J Haematol. 2015;171(5):836 [MEDLINE]